Sampling Methods of Histopathology for Fish and Shellfish  日本語版
日本語版
The histological methods for paraffin sections were established in the 19th century and their fundamentals remain unchanged to date. Although the technique is old, it is still widely used in life sciences, including the state of the art molecular biology, such as the observation of histological changes in genetically modified animals. In clinical medicine, histopathological diagnoses are routinely carried out with biopsy or surgical specimens in medical centers. Although histological methods for fish and shellfish do not depart substantially from those used for humans or mammals, the correct sampling and fixation procedures are crucial to successful histopathological investigation of aquatic animal tissue. Special considerations for aquatic fauna histopathology are discussed in more detail subsequently. The aim of this article is to explain how to sample and fix tissues of aquatic animals, particularly farmed fishes and shellfishes.
1. Characteristics of histopathology
Compared with the other diagnostic methods, histopathology has following characteristics, particularly for fish and shellfish.
- (1) The amount of information obtained from histopathology can be much greater than that from other biochemical, bacteriological, or virological methods.
- (2) Etiological microorganisms that cannot be detected by culture or biochemical methods can be sometimes detected by histopathology. For example, the diseases (etiological agents) we have found so far in our lab primarily by histopathology include: red seabream iridovirus (Fish Pathol. 1992, 27, 19-27), white spot syndrome of shrimps (Fish Pathol. 1994, 29, 135-139), francisellosis in abalone (Dis. Aquat. Org. 2010, 89, 145-154), soft tunic syndrome of tunicate (Dis. Aquat. Org. 2010, 90, 229-240; Dis. Aquat. Org. 2011, 95, 153-161), and encephalomyelitis in amberjack (J. Fish Dis. 2011, 34, 901-910).
- (3) Even when a certain microorganism is detected by Polymerase Chain Reaction (PCR) techniques or isolated by bacteriological or virological methods, it is often difficult to determine whether the organism is the primary causative agent for the disease, particularly when the disease is unknown currently. On the other hand, one can sometimes conjecture with fair certainty whether or not the detected organism is actually the causative agent by observing the organism in situ histologically.
- (4) When the etiological microorganism is culturable, the sensitivity of histopathology to detect the organism is usually very much lower than the ordinary bacteriological or virological methods, not to mention PCR.
- (5) The quality or the amount of information obtained through histopathology varies greatly depending on the observer’s skill. Preparation of histological sections and acquiring information from the sections are two different things. In this regard, histology is different from many biochemical methods, by which you can obtain information if only you learn the procedures.
Important notes for the sampling of farmed fish or shellfish for histopathology
It is true that basic techniques of histology are principally the same for mammals (including humans) and aquatic animals. For aquatic animals, however, several points that are unique to these organisms require particular attention (especially farmed aquatic animals). Usually the death of one or two individuals in a group of farmed aquatic animals is not a problem. Instead, diagnosis is almost always requested only when mortality is high. Therefore, the purpose of diagnosis is to find out if there is a common disease underlying the high mortality and if so, to identify the cause of the disease. Furthermore, unlike clinical medicine for humans or other mammals, for which diagnosis usually means an attempt to classifying an individual’s condition to a known (already designated) disease, researchers often encounter new and unknown diseases in fishes and shellfishes. This makes histopathology especially important for the diagnosis of these animals.
1. For farmed aquatic animals, the subject of diagnosis is usually a group of the animals cultured in a net pen or a tank, and not a particular individual. Therefore, multiple specimens should be sampled from the group. However, the labour-intensive nature of histopathology means that we usually ask people to submit 5-10 specimens.
2. The animals that show typical clinical signs should be sampled. If this is difficult, at least it must be recorded how specimens are
sampled, because the diagnosis may change by how and what kind of specimens
are used. For example, suppose there is daily mortality of 2% in a net
cage that contains 5000 animals and moribund fish exhibit a common clinical
sign before death. This means 100 deaths a day, which is not a small number.
Now, suppose you sample 10 specimens that clearly show the clinical sign
from the cage and find a serious histopathological change in 2 specimens.
Can you say the mortality is caused by the disease that is characterized
by the histopathological change? The answer is no.
Because all subjected specimens showed a typical clinical sign, the causative
lesion should be observed in most of the samples. On the other hand, there
are cases when you cannot sample animals showing typical disease signs
and you have to sample specimens at random. If you find a lesion that is
common to 2 specimens out of 10 randomly sampled fish, the ratio is 20
%. This seems enough to cause the observed mortality of 2 % a day. Thus it is appropriate to consider the lesion as likely to be directly
related to the mortality.
Case #1
A request for diagnosis came from a local fisheries research lab in the southern part of Japan. In a fish farm in the region, many flounder (Paralichthys olivaceus) in farming tanks showed abnormal, peculiar swimming behavior and died. The cumulative mortality reached 20% within a month. Fixed tissue samples from 5 specimens that showed typical strange swimming behavior were sent to the NRIA. In the brain of one individual, many cysts of a myxosporean parasite (probably Kudoa yasunagai) were observed, which seemed to explain the clinical picture well. However, no myxosporean parasite was observed in the other 4 specimens. The researchers at the local fisheries research lab also looked at wet mounts of the brains of 10 affected fish and found the myxosporean cysts from only 2 of them. The parasite at the other developmental stages was not observed even with in situ hybridization. There was no clear inflammatory response around the cysts in the brain. Thus, the possibility of myxosporean etiology was dismissed. Neither other possible causative agent nor lesions were found in the observed specimens, and the cause of the disease remained unknown.
Case #2
Substantial deaths were found in an oyster farm. Although spat of the same
origin were distributed to different farms, a high mortality rate was observed
in only one of them. The mortality reached 10% within a month. Researchers
in the local fisheries research lab found a certain kind of ciliate in
the fresh gill samples of 2 oysters among 6 oysters they observed. Since
it was impossible to choose affected oysters, 14 oysters were randomly
selected and the tissues were fixed. Histological lesions were found in
3 of the 14 oysters. The lesions were principally the same in the 3 specimens.
The epithelial cells of the gills and the edge of the mantle were often
dead or sloughed off in large areas. Dead cell debris and, many, presumably
migrating cells, were found in the lesions. Many ciliates were found both
outside and inside of the lesion. No other notable histopathological changes
were observed in these 3 oysters. Neither histopathological lesions nor
ciliates were observed in the other 11 oysters. It was concluded that the
mortality was caused by the damage inflicted by the ciliate.
3. When a disease occurs and it might be hitherto unknown, diseased specimens should be sampled in an early phase of the disease outbreak. The samples should include fixed tissues for histopathology, frozen animals or tissues for the isolation of pathogens or experimental infections, and tissues preserved in 100% ethanol for gene analyses. This is because detection of the pathogen usually becomes very difficult as the disease subsides, although this is often the case, unfortunately. When an unknown disease occurs, researchers in local fisheries research labs often struggle for a while to identify the etiology by themselves in vain. After that, they request diagnosis from the NRIA, at which time the disease has already begun to subside.
4. Tissue samples should be taken from live animals unless you can obtain only dead samples. Compared with mammals, autolysis (self-digestion) after death proceeds very rapidly in most finfish and aquatic invertebrates. Although it depends on the species or storing conditions, tissues of fish or aquatic invertebrates that have been dead for 30 minutes are usually already inappropriate for histology. You should keep animals alive and subject them for tissue sampling, one individual at a time. If the lab is far away from the place where the disease is occurring, you should take dissecting tools and fixative there to fix tissues at the scene.
Case #3
A disease occurred in yellowtail (Seriola quinqueradiata) cultured in offshore net pens. The size of the fish was 35-40cm in total
length. The fish farm was a few hours’ drive from the nearest lab. Because
affected fish showed strange swimming behavior (suggesting the abnormality
in the central nervous system), the brain had to be sampled. This meant
it would take about 20 minutes to sample tissues from one individual specimen.
We borrowed a room in the office of the fishermen’s cooperation, but it
seemed still impossible to keep moribund fish alive in a small tank while
sampling (we planned to fix tissues from 10 specimens). We brought 10 live
specimens that showed typical strange swimming behavior from a net pen
to the office, and put the fish into a tank filled with seawater and seawater
ice to lower the body temperature of the fish so that the postmortem autolysis
would be delayed. One person then bled the fish by syringes and put them
back in the ice-cold seawater, and I performed tissue sampling for histology
from the fish one by one. The tissue morphology was preserved fairly well.
Case #4
High mortality occurred in wild ayu (Plecoglossus altivelis) in a river in the western part of Japan. A request for diagnosis was
made by a researcher in the nearby local fisheries research lab to the
NRIA. Before sampling, the researcher consulted us about the tissue sampling.
The person was not familiar with histology and it was difficult for her
to conduct tissue sampling at the river. It was also expected that moribund
fish would die while being transported to the lab. I asked her to put moribund
ayu in ice-cold water and bring them to the lab to dissect fish and fix
tissues. In the histological sections, although part of the intestine was
disintegrated by autolysis, morphology of the other tissues was preserved
rather well so that the histological observation was possible. Histopathology
revealed that all specimens had severe inflammatory lesions with numerous
bacterial cells, and hence it was concluded that the disease was a bacterial
infection. The bacterium was identified as Edwardsiella ictaluri by bacteriological and biochemical analyses.
Case #5
There was a request for diagnosis from a researcher of a local fisheries
research lab to the NRIA. He had already sampled fish when he made the
request. He took moribund fish (rainbow trout, 6-7cm in total length) at
the fish farm and tried to bring the fish alive to the lab, although the
fish all died on the way to the lab. He nevertheless made a longitudinal
incision in the abdomen of each specimen and put the whole body of fish
into the fixative. In the histological sections, however, the visceral
organs such as the intestine, pancreas, and liver of most of the fish were
found to be liquefied or disintegrated by autolysis. In some cases, the
muscular tissue of the abdominal wall was also liquefied. Thus, the diagnosis
was impossible. The researcher was apparently not familiar with histological
methods. If only he had consulted us before sampling....
Case #6
Diagnosis was requested by a public fish hatchery for flounder (Paralichthys olivaceus) that showed a markedly high mortality. The fish had been fixed already when the request came to the NRIA. Fish were small (less than 1.5 cm in total length) and the whole body was immersed in the Davidson’s fixative. When the histological slides were observed, the visceral organs of 7 out of 10 specimens subjected for the histology were found to be disintegrated. This means that these fish had already been dead when sampled, because, if the flounder at this size is alive when it’s fixed in the Davidson’s solution, tissue morphology is usually well preserved even when the whole body is immersed in the fixative. Indeed, the tissues of the other 3 specimens were preserved well. The person in charge was not familiar with histology and he apparently didn’t know the need to sample live fish. I wish he had consulted us before sampling. In this particular case, however, the observable 3 specimens had multiple syncytia in the liver or in the intestinal epithelium with extensive cell deaths or sloughing of the epithelium. Thus, the disease was diagnosed as a reovirus infection.
5. Tissues should be sampled according to the lesions or clinical signs. When an unknown disease occurs, and no particular clinical signs are observed, the brain, gill, heart, kidney, and visceral organs should be sampled for histology. When the neuronal disorder was suspected, particularly when affected fish show an abnormal behavior, the brain (and perhaps the spinal cord) must be included in the samples.
Usually, the skin and lateral muscle are not sampled, but if clinical signs are observed there, those tissues should be sampled so that the abnormal portion is included in the sample. This sounds so obvious that there seems no need to mention it, but there are actually cases that a person samples only the tissues of internal organs and doesn’t sample the tissue (such as skin) where lesions are observed clinically. When a whole lesion cannot be sampled because it’s too large, a part of the lesion should be dissected and fixed. In such a case, it is important to include the boundary of the normal tissue and the lesion in the dissected tissue block, because the boundary is the place where the nature of the lesion is apparent and the causative agent, if any, is most often found.
Case #7
There was a request for diagnosis of farmed grouper. Significant mortality had been occurring for months and affected fish were severely emaciated. Researchers at the local fisheries research lab had found a myxosporean parasite (Enteromyxum leei) in the intestine of the diseased fish, and hence, the fish had already been diagnosed as myxosporean emaciation disease when the request was made. However, they had also isolated Photobacterium damselae and Vibrio sp. from diseased fish and the color of the livers of diseased fish was
often dark green. The person in charge called me before sampling tissues,
and I asked her to take the common bile duct and fix it along with the
other tissues. The histopathological observations confirmed the presence
of myxosporean parasite in the intestinal epithelium, which was often sloughed
off into the lumen. The myxosporean parasite was also observed in the epithelium
of the common bile duct, where sloughing of the epithelium and severe inflammation
containing large bacterial masses often filled the duct, causing the biliary
obstruction. Thus, the color of the liver turned green by the accumulated
bile that couldn’t be excreted. In the liver, there were also abscesses
and granulomas apparently caused by bacteria that entered into the liver
through bile ducts. In this case, it would have been impossible to explain
the presence of bacteria, greenish color of the liver, and the lesions
formed in the liver, if the bile duct had not been sampled.
2. Tissue Fixation
General considerations
It is impossible to overemphasize the importance of fixation. Many people
may think it is O.K. to simply put tissues or fish body into a fixative.
On the contrary, proper tissue fixation for histology requires knowledge
and experience. If you fail to fix tissues properly, it can never be compensated by subsequent processes. On the other hand, if tissues are fixed properly, slide preparation is almost guaranteed to be successful.
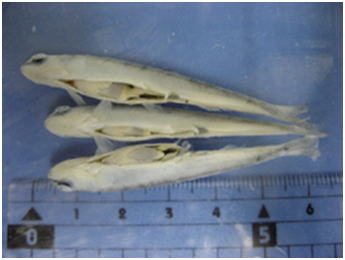
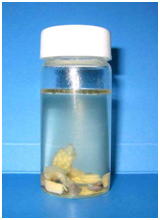
Tissues of fish or aquatic invertebrates are rapidly degraded by autolysis after the death of the organism, and hence, the key to good fixation is how quickly you can inactivate the endogenous enzymes that are responsible for self-digestion. Postmortem autolysis is especially quick in the brain and intestine. There are active digestive enzymes in the intestinal lumen particularly when there is ingested food. Therefore, care should be taken so that the fixative directly enters the lumen of the intestine. For small fish, some people just make an incision on the abdomen and immerse the whole fish body in fixative (Fig. 1). The results of such fixation are very poor (Figs. 2 and 3) even for small experimental fishes like the zebrafish or medaka.
You have to dissect out and trim tissue blocks so that the fixative permeates
target tissues quickly. On the other hand, if the fixed tissue piece is
too small, subsequent handling is difficult. Usually, the thickness of
a dissected piece of an organ or a tissue cluster for fixation should not
exceed 5mm. Thus, however large the animal is, there is a limit for the
size of the pieces of tissues that can be fixed for histology. If you sample
many tissue pieces from each organ, the sensitivity and specificity of
diagnosis for that particular specimen increase, but you should rather
use your time and effort to increase the number of animals you sample for
the diagnosis of farmed aquatic animals, for the reasons described above.
All tissues from one individual specimen are usually fixed in a single
bottle. The volume of the fixative should be more than 10 times (preferably
more than 20 times) the total volume of the tissues that are fixed (Fig. 4).


 |
Fig. 5 Dissecting tools for sampling tissues from small fish. 1, Curved iris scissors; 2, Crown scissors; 3, Fine tweezers; 4, Curved fine tweezers; 5, Scalpel handle (No.4); 6, Tissue forceps; 7, Curved larger tweezers; 8, Scalpel blade (#24; the longer the better). For larger animals, a large knife, kitchen scissors, and a cutting board are useful. You also need; ・An anesthetic ・Paper towels ・Fixative in sample tubes (preferably cooled on ice) |
A sampling sequence for small fish
Although sampling procedures for tissue fixation should vary depending on the animals and situations, an example is described here for small fin fish. In general, the skin and gills should be sampled first, before being dried or damaged by handling. Then, the visceral organs are sampled. Subsequently, the kidney and heart are sampled. Lastly, the brain is sampled, since the brain is covered with a cranium and it often takes time to sample the organ. Agitate the fixative briefly, at each time you put a piece of tissue into the fixative.The overall sampling should be carried out as quickly as possible, since postmortem autolysis proceeds even while you are dissecting the fish
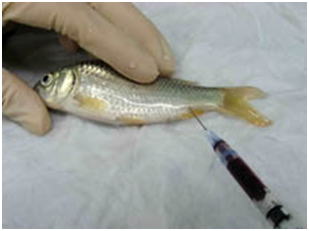 Fig. 6 Fish is anesthetized and the blood is withdrawn from the caudal vein/artery as much as possible. (carp) |
- (1) Wet several overlapping soft paper towels, and wring them so that the water doesn’t drop. Do not wring them dry. Anesthetize a single specimen from the group of fish you are going to sample.
- (2) Place the fish on the wet paper towels and draw blood from caudal artery/vein
(Fig. 6) as much as possible. Alternatively, you can bleed the animal by severing
the caudal peduncle or ventral aorta.
Otherwise tissue sampling would be hampered by blood when you open the abdominal cavity. Furthermore, blood often interferes in fixation process or in observations of tissue sections. It is also difficult to identify blood congestion, if the blood has not been removed. In addition, blood causes the deposition of formalin pigment, if you use formalin as part of the fixative without neutralization. You can make blood smear preparations here, if necessary. - (3) Remove a few gill arches and put them in the fixative. If there is a macroscopic lesion on the skin, the portion should be sampled together with the underlying muscle and fixed (Figs. 7).
- (4) Open the visceral cavity as widely as possible to sample visceral organs.
For a small fish, the cluster of whole visceral organs should be removed
by severing the digestive tract at the esophagus and anus. Do not separate
organs from each other (Fig. 8). Hold the organ cluster with forceps on wet paper towels and cut it by moving the blade of a scalpel between the tips of the forceps. Move the blade rather horizontally. Do not compress tissues with the blade. Cut out a 5mm-thick tissue block (Fig. 9) by cutting the visceral organs transversely.
Be sure that the tissues you need are included in the block. Cut out multiple tissue blocks if necessary. In this way, the fixative enters the lumen of the digestive tract quickly. This is important since the post-mortem autolysis proceeds very rapidly in the epithelium of the digestive tract where active digestive enzymes are often present. If the fish is large, a piece of tissue is sampled from each organ. For such a case, a piece of intestine should be less than 1 cm for fixation. Do not remove attached tissues such as the mesentery. Lesions often develop on the mesentery or in the intraperitoneal fat. Also, the pancreas is scattered in the mesentery. If the fish has many pyloric caeca, a bunch of them should be cut out into 5mm-thick block by cutting perpendicularly to the caeca without separating each one of them, so that you can observe the mesentery, fat, and pancreatic tissues among the caeca.
- (5) If there are any lesions, such as hemorrhage, in the lateral muscle, a piece of muscle should be sampled including the lesion. The muscle is usually cut out as a 5mm-thick section perpendicular to the muscle fibers. If the fish is small, the lateral muscle should be sampled with skin. To cut out the muscle, only scalpels or razor blades should be used. Avoid using scissors, which may destroy substantial part of the dissected musculature.
- (6) Fix the body kidney (Fig. 10). If the fish is large, a piece of the tissue can be cut out from the organ. If the fish is small, the whole body kidney should be fixed along with some muscle and vertebrae surrounding the kidney. Further trimming can be done after fixation.
- (7) Remove the heart (atrium, ventricle, and bulbus arteriosus) and put it in the fixative. If possible, the heart should be cut into two pieces along the midsagittal plane so that the lumen of the ventricle and atrium comes in direct contact with the fixative.
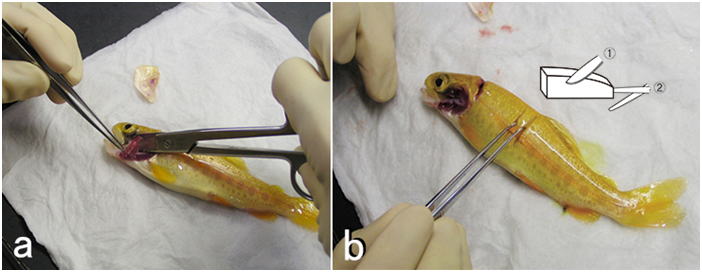
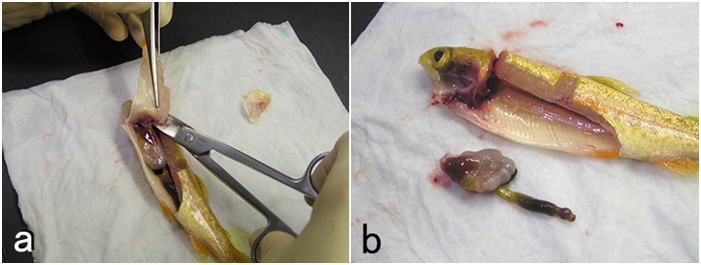
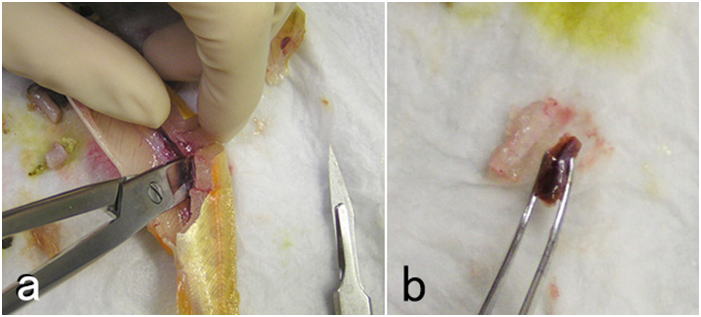
- (8) Sever the head from the body at the posterior end of the operculum. Remove the lower jaw. Remove tissues surrounding the cranium as much as possible. Carefully make a small opening in the cranium and expose part of the brain (Fig. 11), so that the fixative enters the brain case through the opening. Now, the brain can be fixed together with the cranium. If the fish is smaller, the whole severed head can be directly placed into the fixative solution, although it is still a good idea to remove the lower jaw from the head since the oral cavity may contain air. If the fish is large, a meat cleaver or a thick-bladed carver for dressing fish is useful. To dissect out the whole brain, remove the muscle and connective tissue attached to the cranium using crown scissors or kitchen scissors as possible as you can. Then, remove the upper and side part of the cranium to expose the brain as much as possible. Hold the posterior end of the medulla oblongata or the anterior end of the spinal cord with a pair of tweezers and lift it gently as you sever brain nerves from posterior to anterior. Remove the brain completely by cutting the optic nerve and olfactory nerve (or olfactory tract). The pituitary is usually left on the base of the cranium. If needed, the brain may be cut into smaller parts before fixation.


If the specimen is very small, such as alevins of trout or larvae of seawater fish, the whole body of fish can be directly immersed in the fixative. Anesthetization is necessary also for such small specimens. Because if fish are fixed without anesthetization, they struggle with agony and fish may be fixed with the body being bent. This is not only cruel but also the orientation of the specimen for paraffin embedding or subsequent sectioning would become difficult.
After fixation
Immediately after putting all necessary tissues of an individual animal into the fixative, the bottle should be shaken slowly on an automatic shaker. The bottle should be set on the shaker horizontally so that the fixative can be well agitated. If you don’t have a shaker, you can agitate the fixative by moving the bottle upside down every few minutes at least for the first 30 minutes after fixation. The interval can be longer as the time passes. After that, you can leave the bottle to stand still. This agitation is very important. If the fixative is not agitated, the fixative surrounding tissues would be rapidly diluted by the body fluid seeping out of the tissues, and fixation would proceed only slowly, allowing autolysis.
The tissue is fixed overnight at room temperature (This is enough for Davidson’s or Bouin’s solution). Then, the fixative is discarded and the tissues are rinsed in 70% ethanol for 1 hour. Subsequently, the 70% ethanol is changed to new 70% ethanol. Then, the tissues can be stored at least for a few weeks (when Davidson’s solution is used).
To decalcify tissues, tris (50mM)-buffered 400 mM EDTA (pH 7.5) is used in our lab, although it takes more time than acid decalcifiers.
Thickness of paraffin sections
In general, histological sections of fish and aquatic invertebrates should be cut at 3-4 μm (preferably 3μm), because cells of these animals are substantially smaller than those of mammals. In thicker sections, the cells often overlap each other and intracellular structure is difficult to observe. Sections at 4-5 μm will do for some fish, such as carp or salmonids, since the cells of these fish are much larger than those of the other teleosts. This is probably because these fishes have experienced tetraploidization rather recently, and the genome size, hence the cell size, is large. When fixation is inappropriate, thin sections tend to look hollow and mesh-like as in the left panel of Fig. 3, perhaps because intracellular substances are lost substantially. In such tissues, thicker sections may look superficially better. To make good histological preparations, however, you have to fix tissues properly, cut thin sections, and stain them intensely. If you plan to take photos of sections at a low magnification, you may cut some thicker sections, so that you can take photos of good contrast at low magnifications.Choice of fixatives: 10% formalin is inappropriate
Davidson’s fixative is recommended for general histopathology of fish and shellfish. Don’t use simple formalin solutions, such as 10% neutral buffered formalin. These fixatives are unsuitable to fix tissues of fish and aquatic invertebrates, in which the postmortem autolysis (self-digestion) proceeds very rapidly. In other words, if autolysis doesn’t proceed rapidly, you can obtain good results with 10% formalin. Although there are indeed such cases even with aquatic invertebrates, it is safer to use Davidson’s solution for general histological studies of fishes and shellfishes. Researchers studying molluscs or crustaceans most often use Davidson’s fixative for histology. It is surprising that many researchers are still using simple formalin solutions to fix tissues of fin fishes. This is probably because no literature clearly states that a simple formalin solution is inappropriate as the fixative for aquatic organisms including fish. Although it is said that formalin permeates tissues rather quickly, it takes 3 hours to fix 95% of the protein in the tissue (Mizuhira, 1990). A simple formalin fixative is often recommended for histology of mammals. Postmortem autolysis of mammals is much slower than that of fish. In the tissues of mammals including human, coagulation necrosis primarily occurs after cell death (Majno and Joris, 2004), whereas liquefaction necrosis seems to prevail in fish after death. If a piece of rat liver is implanted in the peritoneal cavity of another rat (thus, without blood supply, the tissue is under the same condition as in an infarct) and fixed after 2 hours, when most of the cells are dead, no pathologist would be able to tell that the cells are dead (Majno and Joris, 2004). On the other hand, if a fish is left at room temperature for 2 hours after death, part of visceral organs would be disintegrated by autolysis. In fish (the visceral organs and brain, in particular) or aquatic invertebrates, autolysis starts immediately after death. In fixatives like Davidson’s or Bouin’s solutions, proteins including various enzymes in tissues are probably denatured rapidly by reagents such as ethanol, acetic acid, or picric acid, and the fixation by formaldehyde proceeds subsequently. When the liver of trout is fixed in either 10% neutral buffered formalin or Davidson’s solution for 35 minutes, a substantial amount of soluble protein still remains in the tissue fixed in 10% formalin, whereas virtually no soluble protein remains in the tissue fixed in Davidson’s fixative (Fig. 12).
Comparison of four fixatives, 10% neutral buffered formalin, Davidson’s fixative, Bouin’s solution, and Helly’s fluid, indicates superiority of Davidson’s fixative for histology of fish in general (Figs. 13-17). Bouin’s solution is a good fixative, although the tissues fixed in Bouin’s solution cannot be stained well with Giemsa (Fig. 18), and hence, inappropriate for general histopathology, since Giemsa stain (we usually use May Grünwald-Giemsa stain) is very useful to detect bacteria, protistan parasites or fungi. Excellent results are sometimes obtained with fixatives containing mercury, such as Helly’s or Zenker’s fixative. These are not recommended, however, because they contain heavy metals, which make the treatment of waste material difficult. Besides, only poor results are obtained when fish larvae are fixed with Helly’s solution (Fig. 17). Additionally, you have to remove the mercury deposition from the sections of tissues fixed in these solutions. Davidson’s solution also has its own drawbacks. The biggest of all is the formation of hematin or “formalin pigment”, which may disturb observations if not treated properly (Fig. 19). Hematin is formed from hemoglobin with formalin under acidic conditions. Thus, hematin is not produced by neutral buffered formalin. Hematin is neither produced in Bouin’s solution, since picric acid dissolves the pigment. Davidson’s fixative contains acetic acid with a high concentration of formalin, providing an ideal condition for hematin formation. In addition, in the organs fixed in Davidson’s solution, the epithelium is often detached from the underlying tissue. This is often observed in the gills (Fig. 20). Another major drawback of Davidson’s solution, as well as Bouin’s fluid, is hemolysis as shown in Fig. 21. In addition, Davidson’s fixative may sometimes be inappropriate for immunohistochemistry (IHC), while the preservation of antigenicity is usually better in the tissues fixed in neutral buffered 10% formalin (Fig. 22). This is probably because many epitopes are denatured or modified by “strong” fixatives. On the other hand, there are cases where 10% formalin is inappropriate, such as for cholecystokinin (CCK) (Fig. 23). For CCK, the antibody reaction was only obtained with the tissue fixed in Bouin’s solution. Since CCK is a peptide hormone, formalin alone was probably not enough to fix it. CCK may have been dissolved out or degraded by autolysis in the formalin-fixed tissue. Davidson’s fixative or Helly’s solution was apparently too “strong” for such a peptide hormone, which has only a limited number of epitopes. I usually obtain good IHC results with tissues fixed in Davidson’s solution, however, to detect pathogenic microorganisms by using polyclonal antibodies raised against the whole body of the organism. For in situ hybridization, quick inactivation of nucleases in cells seems also important for fish. I tested the 4 fixatives mentioned above for in situ hybridization of pepsinogen mRNA in the flounder stomach and obtained excellent results with Davidson’s and Helly’s fluid, good results with neutral buffered 10% formalin, but only very weak signal with Bouin’s fluid. On the other hand, I obtained excellent results with the same in situ hybridization system when I used a paraffin block of the flounder stomach fixed in Bouin’s solution prepared in another lab. The reason for this difference is not certain. Although the fixatives were prepared according to the same recipe, the chemicals produced by different manufacturers may affect preservation of nucleic acids differently. In any case, you have to test different fixatives by yourself for these special techniques.











| Methods to compare 4 different fixatives The animals used for the study were; larvae of the Japanese flounder, Paralichthys olivaceus, just after the start of eye migration (n=20), weighing 10-20 mg, juvenile Japanese flounder weighing 214-254g (n=4), and rainbow trout, Oncorhynchus mykiss weighing 123-129g (n=3). Juvenile and larval flounder were purchased from a private hatchery, and transported to the National Research Institute of Aquaculture (NRIA, Nansei, Mie, Japan). The flounder larvae were reared at 20 ℃ by feeding newly hatched Artemia nauplii. The juvenile flounder were reared at 20 ℃ by feeding commercial pellet diet. The rainbow trout were obtained from Nagano Prefectural Fisheries Research Station, and reared in the Inland Station of NRIA (Tamaki, Mie, Japan) at 15-16 ℃ by feeding commercial pellet diet. Lillie’s neutral buffered 10% formalin (NBF) was prepared according to Kiernan (2003), using commercial formalin, which contains 37% formaldehyde and 8% methanol. Bouin’s fluid was prepared by mixing 75 ml of saturated aqueous picric acid, 25 ml of formalin, and 5 ml of glacial acetic acid. Davidson’s fixative was prepared by mixing 33 ml of ethanol (95%), 22 ml of formalin, 11.5 ml of glacial acetic acid, and 33.5 ml of distilled water (Bell and Lightner, 1988). Helly’s fixative was prepared by adding 5 ml of formalin to 100 ml of the stock solution which contained 5g of mercuric chloride (HgCl2), 2.5 g of potassium dichromate (K2Cr2O7), and 1 g of sodium sulphate (Na2SO4.10H2O). NBF was prepared 24 hours before tissue fixation. Other fixatives were prepared within 4 hours before tissue fixation. The fixation was carried out at room temperature (20-25 ℃). Juvenile flounder: Each individual fish was anesthetized with 1/5000 2-phenoxyethanol and bled from the caudal peduncle with a syringe. The brain, intestine, and kidney were excised from each fish, and fixed in the four different fixatives. Thus, 16 sample bottles of fixatives were used in total. The brain was cut along the midsagittal plain, and fixed in two different fixatives; either NBF and Davidson's fixative, Davidson's fixative and Bouin's fluid, Bouin's fluid and Helly's fixative, or Helly's fixative and NBF. The other tissues were further cut into four small pieces of approximately the same size with the thickness of 3-4 mm, and put into the four different fixatives. Each bottle contained 40 ml of fixative and the wet weight of the tissues fixed in each bottle was 0.62 ± 0.03 g (mean ± SEM). Larval flounder were anesthetized with 1/5000 2-phenoxyethanol and five fish were fixed in each of four fixatives. The whole fish body was directly immersed in the fixatives in sample bottles, each containing 45 ml of fixative solution. Rainbow trout (n=3): Each fish was anesthetized with 1/5000 2-phenoxyethanol and bled from the caudal peduncle with a syringe. The brain, pyloric caeca with pancreas, and kidney were excised from each fish, and fixed in the four different fixatives. The tissues were fixed as described for the juvenile flounder. Thus, 12 sample bottles of fixatives were used in total. Each bottle contained 40 ml of fixative and the wet weight of the tissues fixed in each bottle was 0.72 ± 0.03 g (mean ± SEM). After putting the tissues or larvae into the sample bottles containing fixatives, the bottles were immediately put on an automatic shaker and gently agitated while fixing. After 5 hours of fixation, the tissues in Helly’s fixative were washed with running well water overnight and transferred into 70% ethanol. The tissues in Bouin’s and Davidson’s solutions were fixed overnight. The tissues fixed in Davidson's solution were washed in 70% ethanol and stored in new 70% ethanol. The tissues fixed in Bouin's solution were washed 3 times with 70% ethanol until most of the yellow color is removed from the tissues. The tissues in NBF were fixed for 3 days and transferred into 70% ethanol. After transfer into 70% ethanol, all tissues or fish were routinely embedded in paraffin. Before staining, deparaffinized sections of tissues fixed in Davidson’s solution were transferred from xylene to absolute ethanol and then to 98% ethanol. Then the sections were immersed in 95% ethanol saturated with picric acid for 5 minutes. Subsequently, sections were briefly washed with water and then placed in 50% ethanol for one minute. Sections were then washed in running water until the yellow color of picric acid disappeared completely. Before staining, deparaffinized sections of tissues fixed in Helly’s fixative were treated in the mixture of 3 volumes Lugol’s solution and 7 volumes 70% ethanol for 10 minutes to remove precipitates of mercury. Sections were then rinsed in water, decolorized in 0.25% sodium thiosulfate for 30 seconds and rinsed in water. Two sets sections were cut at 3 μm from each tissue. One set was stained with Mayer’s hematoxylin and eosin (eosin Y and erythrosine). The other was stained with May-Grünwald and Giemsa. For this staining, deparaffinized sections were stained in May-Grünwald solution (10 ml of commercial May-Grünwald solution and 40 ml of distilled water) for 3 hours and in Giemsa solution (Wolbach method) overnight. The sections were then differentiated in acetic acid (one or two drops of 100% acetic acid in 100 ml distilled water), and dehydrated through acetone. For the kidney of the juvenile flounder and trout, 2 μm-thick sections were prepared and stained with Azan. Immunohistochemistry (IHC) for tripsinogen in the pancreatic tissue and for cholecystokinin (CCK) in the intestine of the flounder was carried out with rabbit anti-eel tripsinogen antiserum (Kurokawa and Suzuki 1995) and rabbit anti-flounder CCK antiserum (Kurokawa et al., 2000), respectively. Sections were cut at 3 μm and deparaffinized. Slides were first treated with normal goat serum diluted to 1/20 with 1% bovine serum albumin (BSA) in 10 mM phosphate buffered saline (PBS) for 20 minutes. Slides were then treated with the primary antibody (1/100 for CCK and 1/500 for tripsinogen) diluted with 1% BSA in 10 mM PBS for 40 minutes. Subsequently the bound antibody was detected using VECSTAIN Elite ABC kit (Vector Laboratories, Inc. Burlingame, CA, USA) with diaminobenzidine tetrahydrochloride and hydrogen peroxide as the substrates. For the control, normal rabbit serum was substituted for the primary antibodies. |
Kiernan, J. A. (2003) Histological & Histochemical Methods: Theory & Practice. 3rd ed. Oxford University Press.
Kurokawa, T. and Suzuki, T. (1995) Journal of Fish Biology 46, 292-301.
Kurokawa, T., Suzuki, T., and Andoh, T. (2000) General and Comparative Endocrinology 120, 8-16.
Majno, G. and Joris, I. (2004) Cells, Tissues, and Disease; Princeples of General Pathology 2nd ed. Oxford University Press.
Mizuhira, V. (1990) Electronmicroscopy 25(1), 44-57. (In the Japanese language)